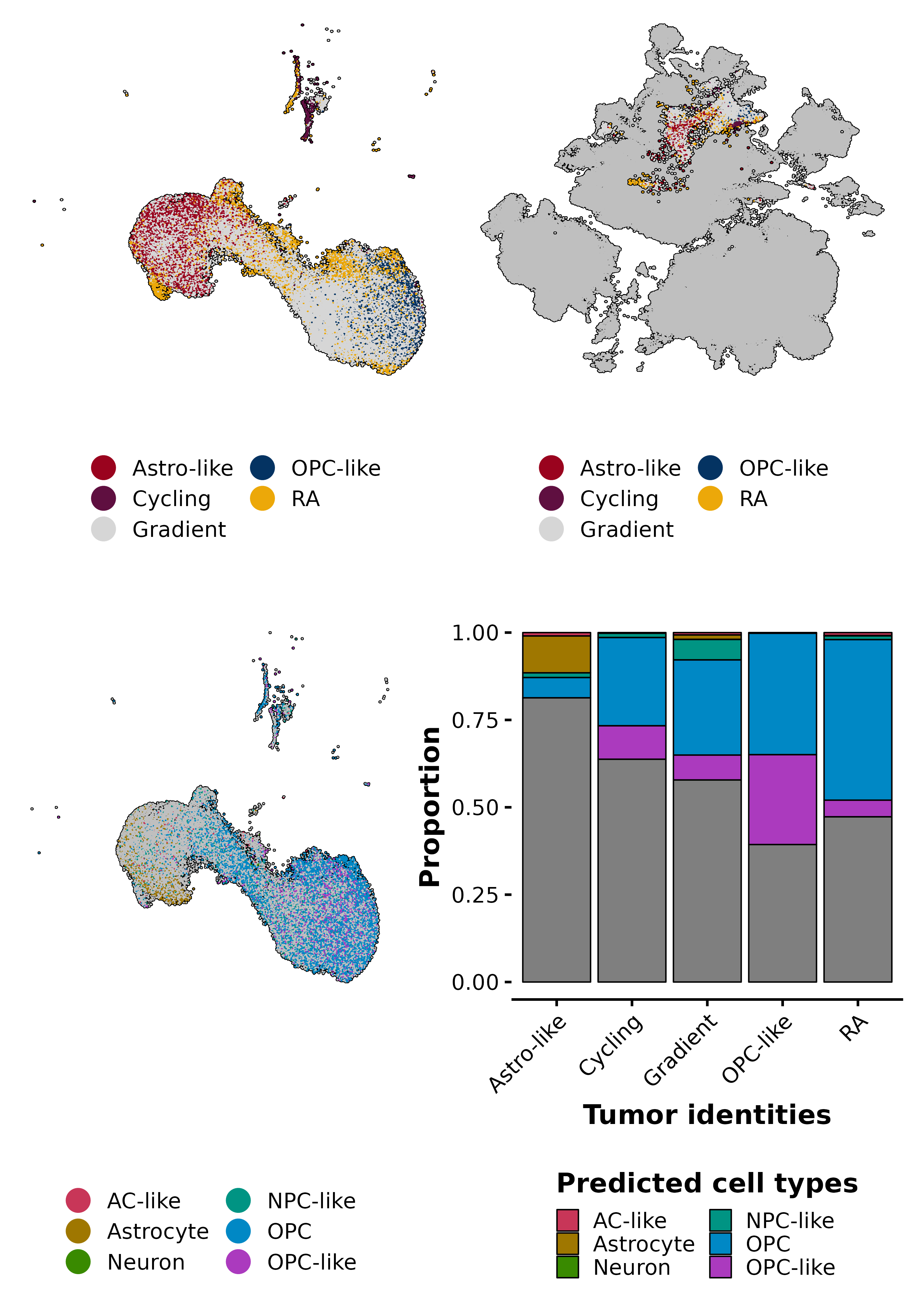
p1 <- SCpubr::do_DimPlot(sample = sample,
reduction = "umap",
colors.use = cluster_cols,
raster = TRUE,
raster.dpi = 2048,
pt.size = 4,
legend.icon.size = 8,
legend.ncol = 2,
font.size = 20)
p2 <- SCpubr::do_DimPlot(sample = sample,
reduction = "ref.umap",
colors.use = cluster_cols,
raster = TRUE,
raster.dpi = 2048,
pt.size = 4,
legend.icon.size = 8,
legend.ncol = 2,
font.size = 20)
# Add reference coordinates.
data <- as.data.frame(azimuth_core@reductions$umap@cell.embeddings)
colnames(data) <- c("x", "y")
base_layer <- scattermore::geom_scattermore(data = data,
mapping = ggplot2::aes(x = .data$x,
y = .data$y),
color = "grey75",
size = 4,
stroke = 4 / 2,
show.legend = FALSE,
pointsize = 4,
pixels = c(2048, 2048))
p2$layers <- append(base_layer, p2$layers)
base_layer <- scattermore::geom_scattermore(data = data,
mapping = ggplot2::aes(x = .data$x,
y = .data$y),
color = "black",
size = 4 * 2,
stroke = 4 / 2,
show.legend = FALSE,
pointsize = 4 * 2,
pixels = c(2048, 2048))
p2$layers <- append(base_layer, p2$layers)
p3 <- SCpubr::do_DimPlot(sample = sample,
reduction = "umap",
group.by = "inferred_annotation",
raster = TRUE,
raster.dpi = 2048,
pt.size = 4,
legend.icon.size = 8,
legend.ncol = 2,
font.size = 20)
p4 <- SCpubr::do_BarPlot(sample = sample,
group.by = "inferred_annotation",
split.by = "relabelling",
position = "fill",
legend.title = "Predicted cell types",
font.size = 20,
axis.text.face = "plain",
legend.ncol = 2) +
ggplot2::xlab("Tumor identities")
p <- (p1 | p2) / (p3 | p4)